
The technologies of Genotyping by Sequencing (GBS) and Restriction site Associated DNA Sequencing (RAD-Seq) have emerged as dual catalysts in the domain of genetic variation detection, reshaping research paradigms from crop breeding to ecological evolution through their high-throughput and cost-effective strategies. This article provides a systematic analysis of the core principles underlying these technologies—GBS employs random enzyme digestion to simplify genomic complexity, whereas RAD-Seq focuses on precise capture of variation signals at restriction sites. Their application scenarios are compared: GBS accelerates marker detection in agricultural genomic selection, while RAD-Seq, with its adaptability to low-quality DNA, is a powerful tool for studying non-model organisms and endangered species. The discussion also addresses common challenges such as detection in polyploid species and experimental automation, offering solutions and insights. Furthermore, a comparative table of technical parameters offers clear insights into their trade-offs concerning data quality and cost-effectiveness. Whether you are an ecologist investigating population genetic structures or a breeding specialist seeking to optimize marker-assisted selection, this article will uncover how to align technological strengths with research needs, enabling you to gain a competitive edge in the "efficiency revolution" of genotyping.
Introduction to Genotyping Technologies
In the extensive examination of genomics research, genotyping technology has become an increasingly significant instrument for the analysis of genetic variation, the comprehension of the mechanism of biological evolution, and the promotion of innovative breeding practices. Specifically, sequencing-based genotyping technologies, notably Genotyping by Sequencing and RAD-Seq, have emerged as the focal point of contemporary research due to their high efficiency and precision. Both GBS and RAD-Seq are simplified genome sequencing technologies that reduce costs by reducing the amount of sequencing data while retaining sufficient genetic information for accurate genotyping. GBS detects polymorphic sites by randomly interrupting genomic DNA, cutting it with restriction endonucleases, and sequencing specific fragments. Conversely, RAD-Seq focuses on the selection of DNA fragments for sequencing in the vicinity of restriction sites, predicated on the principle that disparities in enzyme cleavage sites between individuals can reflect genetic variation. The emergence of these two technologies is not only attributable to the rapid advancement of high-throughput sequencing technology but also closely associated with the pressing need for comprehensive exploration of genetic information in the context of biological research.
Core Principles of GBS and RAD-Seq
GBS and RAD-Seq are two genotyping techniques that have recently attracted significant attention from the research community due to their unique advantages. The subsequent discussion will provide an exhaustive introduction to the fundamental principles of each of the two techniques.
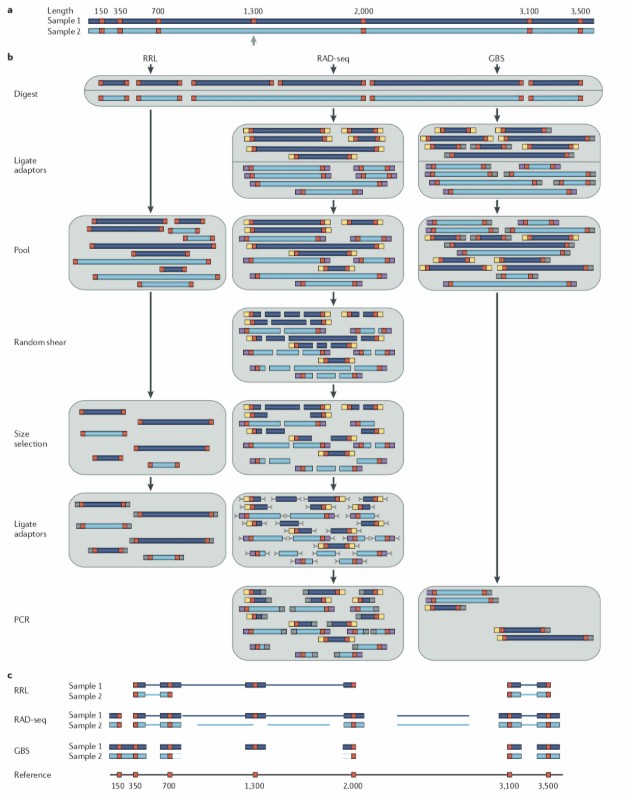
Summary of GBS and RAD-Seq library construction methods (Davey et al., 2011)
Sequencing-based genotyping (GBS)
The fundamental principle of GBS technology is predicated on its distinctive protocol and workflow. Initially, the selection of restriction endonucleases is of paramount importance, directly impacting genome coverage and the efficiency of polymorphism discovery. It is important to note that different enzymes exhibit different cutting site preferences, and as such, researchers must select the appropriate enzyme according to the research purpose and species characteristics. Secondly, GBS optimizes cost-effectiveness through a high-sample multiplex sequencing strategy, where DNA fragments from multiple samples are mixed and sequenced, thus reducing the cost of a single sample. Furthermore, GBS technology exhibits compatibility with sequencing platforms such as Illumina and NovaSeq, thereby enabling flexible selection of sequencing protocols according to the varying project scales.
Within the GBS workflow, the sample DNA is subject to an initial cleavage step, followed by ligation for splicing. The DNA is then subjected to a PCR amplification process, after which the library is formed. Subsequently, the library is transferred to the sequencing platform for sequencing, and the resulting sequencing data is analyzed by bioinformatics, including quality control, comparison, and variant detection, to obtain the genotype information of the sample. This process is not only efficient and accurate but also highly reproducible, providing a strong guarantee for large-scale genotyping research. Mascher et al. conducted a study on the application of sequencing-based typing on semiconductor sequencing platforms and made comparisons between the sequencing of genetic and reference base markers. They concluded that the GBS approach applies to a range of platforms.
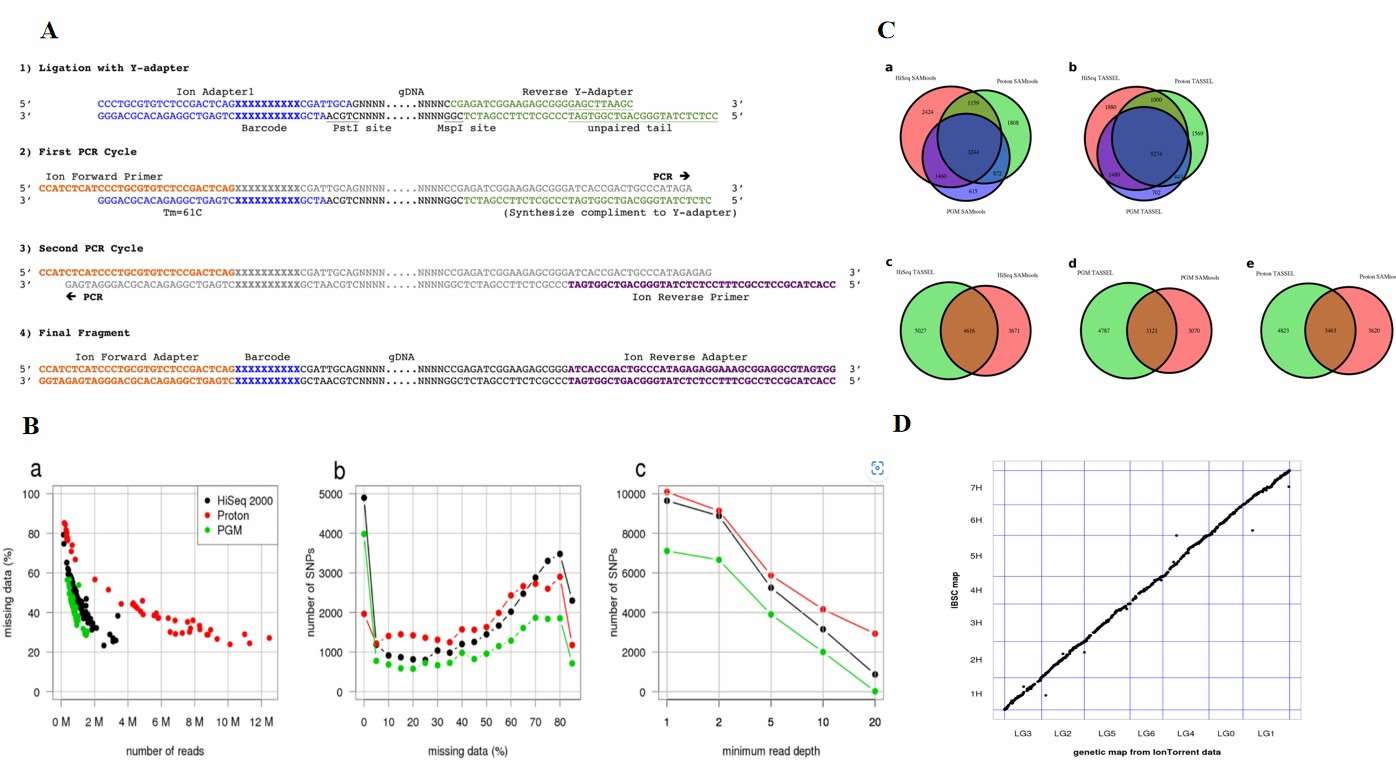
Application of Genotyping-by-Sequencing on Semiconductor Sequencing Platforms (Mascher et al., 2013)
Restriction Site Associated DNA Sequencing (RAD-Seq )
Conversely, RAD-Seq technology facilitates genotyping through the sequencing of DNA fragments in the proximity of restriction sites. In comparison with GBS, RAD-Seq provides a greater range of options in terms of dual-enzyme cleavage versus single-enzyme RAD strategies. The double-enzymatic cleavage strategy has the potential to further reduce genomic complexity and enhance sequencing efficiency; however, it may compromise marker density. Conversely, the single-enzyme RAD strategy, while retaining greater genetic information, is associated with comparatively high sequencing costs. Therefore, it is incumbent upon researchers to evaluate the relationship between complexity reduction and marker density according to the specific aims of the research and the available budget.
Furthermore, RAD-Seq technology emphasizes the selection of fragment size. The choice of fragment size is pivotal in achieving a balance between genomic representativeness and sequencing depth, thus ensuring the accuracy of the sequencing data and concomitantly reducing the costs of the process. Furthermore, RAD-Seq solutions exhibit a high degree of flexibility, with customizable junction designs that enable adaptation to different species-specific applications. This feature confers a unique advantage to RAD-Seq in non-model organism research. Zhang et al. utilized RAD-Seq to capture the variable region of 16S rRNA and the protein-coding genes adjacent to it. This approach integrates classical 16S rRNA amplicon sequencing and macro genome sequencing to address the inconsistency between the two in taxonomic and functional annotation.
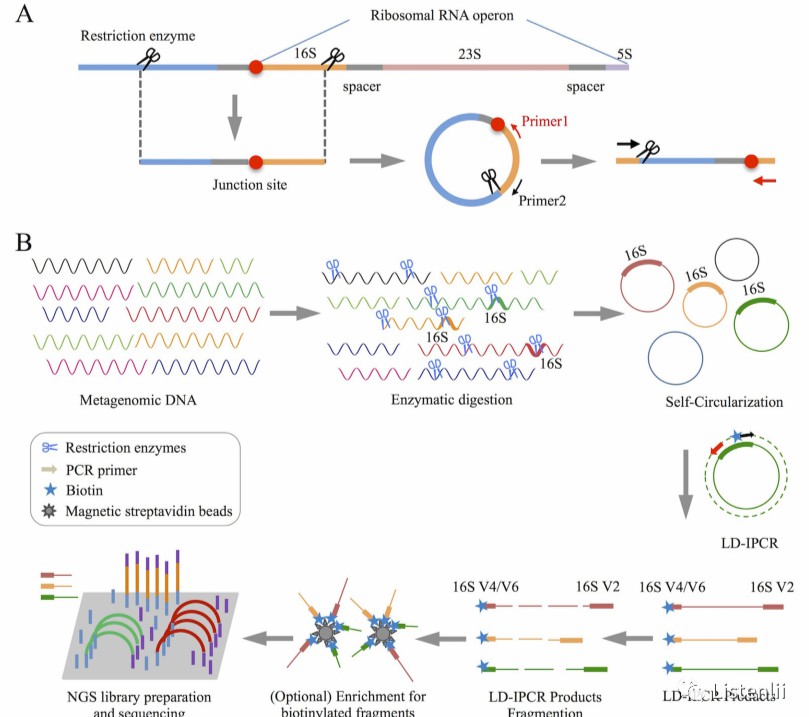
The variable region of 16S rRNA and its flanking protein-coding genes were captured using RAD-Seq (Zhang et al., 2016)
Comparative Analysis: GBS vs RAD-Seq
Genotyping technologies are an essential component of modern biology, and two such technologies of particular note are GBS and RAD-Seq. These technologies differ in numerous ways, and thus, they are suitable for different application scenarios. The subsequent comparative analysis will address the technical distinctions between these two technologies and explore their respective application scenarios.
Technical differences
GBS and RAD-Seq have their advantages and disadvantages in terms of the complexity of library preparation; GBS library preparation is relatively simple, takes less time to perform, requires less technical expertise, and is easily automated. RAD-Seq library preparation, on the other hand, can involve more steps and more complex operations, requiring more technical expertise and laboratory conditions. However, as the technology continues to evolve, the library preparation process for RAD-Seq is being optimized, and its potential for automation is beginning to emerge.
In terms of variance resolution, GBS and RAD-Seq face different challenges in polyploid species. For example, in complex genomic species such as hexaploid wheat, GBS may struggle to detect SNP loci due to high genome complexity. RAD-Seq, on the other hand, may be able to improve variant resolution by optimizing digestion strategies and fragment size selection. Therefore, when choosing a genotyping technique, researchers must fully consider the genomic characteristics of the species and the research objectives.
Application Scenarios
In population genomics research, RAD-Seq is favored for its high marker density and ability to analyze fine structures. RAD-Seq technology enables researchers to reveal the genetic diversity, population structure, and evolutionary history of species. GBS, on the other hand, plays an important role in crop breeding and genetic improvement due to its ability to construct large-scale diversity panels. Using GBS technology, researchers can quickly and accurately determine genotypic information from a large number of samples, providing strong genetic support for crop breeding.
For non-model organism research, RAD-Seq's adaptability to low-quality DNA gives it a unique advantage. For example, in samples with poor DNA quality, such as museum specimens, RAD-Seq can still provide sufficient genetic information for genotyping. GBS, on the other hand, may struggle to produce accurate results due to poor DNA quality.
GBS has received a lot of attention in breeding programs for its ability to provide rapid marker-assisted selection. With GBS technology, breeders can speed up the breeding process by quickly screening individuals with superior traits in early generations. RAD-Seq, on the other hand, can limit its use in breeding programs due to the high cost and complexity of the process.
Challenges and Solutions for GBS and RAD-Seq
The practical application of GBS and RAD-Seq presents several complex issues and challenges, and the common pitfalls and corresponding optimization strategies are detailed below.
Common pitfalls
When using GBS with RAD-Seq, researchers may face challenges such as allele deletion, missing data, and reference genome dependence. Allele deletions can be caused by enzyme bias, where certain enzymes are less efficient at cleaving certain sequences, resulting in some alleles not being detected. To mitigate this problem, researchers can choose enzymes with higher cutting efficiency or optimize the enzymatic conditions. Missing data, on the other hand, can be caused by incomplete loci due to uneven digestion in GBS. To overcome this problem, researchers can adopt stricter quality control standards and data-filling methods. Reference genome dependence, on the other hand, is the main challenge facing RAD-Seq in ab initio SNP discovery in reference-free species. To overcome this, researchers can adopt de novo assembly strategies or use reference genomes from closely related species for assisted analysis.
Optimization strategies
To balance the depth of coverage in polyploid and diploid systems, researchers need to choose the appropriate sequencing depth based on the genomic characteristics of the species and the purpose of the study. In polyploid species, a higher sequencing depth may be required to accurately detect SNP loci. In diploid species, a lower sequencing depth may be appropriate to reduce costs. In addition to modifying the RAD-Seq protocol to accommodate phylogenetically diverse samples, researchers can adjust parameters such as digestion strategy, fragment size selection, and junction design according to sample characteristics and research needs.





